flowchart TD A[Raw counts] --> B[edgeR preprocessing] B --> C[TMM normalization] C --> D[Exploration and QC] D --> E[Design matrix] E --> F[voom transformation] F --> G[Linear modeling] G --> H[Differential expression] H --> I[Visualization and interpretation]
rnaseq-with-replicates-limma
TipReference & License
This tutorial is an independent reimplementation of the Bioconductor RNA-seq workflow for educational purposes.
Original workflow:
- Charity Law, Monther Alhamdoosh, Shian Su, Xueyi Dong, Luyi Tian, Gordon K. Smyth and Matthew E. Ritchie
- RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR
Important
本流程中,edgeR 主要負責處理原始計數資料,包括資料結構(DGEList)、低表現基因過濾(filterByExpr)以及 TMM 標準化,確保樣本之間的 library size 與組成偏差被妥善校正。
完成這些前處理後,voom 會將標準化後的計數資料轉換為 log2-CPM 並估計平均值與變異數之間的關係,進而為每一筆觀測值分配精確的權重。這個步驟使 RNA-seq 資料能夠合理地套用 limma 的線性模型與經驗貝葉斯方法,同時保留對低表現與高變異基因的適當不確定性估計。
1 分析流程圖
2 下載範例資料
以小鼠乳腺 RNA 定序資料為範例,先下載之後解壓縮
url <- "https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE63310&format=file"
utils::download.file(url, destfile="data/GSE63310_RAW.tar", mode="wb")
utils::untar("data/GSE63310_RAW.tar", exdir = "./data")
files <- c("GSM1545535_10_6_5_11.txt", "GSM1545536_9_6_5_11.txt", "GSM1545538_purep53.txt",
"GSM1545539_JMS8-2.txt", "GSM1545540_JMS8-3.txt", "GSM1545541_JMS8-4.txt",
"GSM1545542_JMS8-5.txt", "GSM1545544_JMS9-P7c.txt", "GSM1545545_JMS9-P8c.txt")
files_path <- paste0("data/", files)
for(i in paste(files_path, ".gz", sep=""))
R.utils::gunzip(i, overwrite=TRUE)txt 中的資料為 EntrezID, GeneLength 與 Count,如下:
EntrezID GeneLength Count
497097 3634 1
100503874 3259 0
100038431 1634 0
19888 9747 0
20671 3130 13 讀取資料
使用 readDGE 可以直接讀取多個 txt 檔,files_path 為檔案位置的 list
library(limma)
library(Glimma)
library(edgeR)
library(Mus.musculus)
files_path
x <- readDGE(files_path, columns=c(1,3))
class(x)
dim(x)[1] "data/GSM1545535_10_6_5_11.txt" "data/GSM1545536_9_6_5_11.txt" "data/GSM1545538_purep53.txt" "data/GSM1545539_JMS8-2.txt"
[5] "data/GSM1545540_JMS8-3.txt" "data/GSM1545541_JMS8-4.txt" "data/GSM1545542_JMS8-5.txt" "data/GSM1545544_JMS9-P7c.txt"
[9] "data/GSM1545545_JMS9-P8c.txt"
[1] "DGEList"
attr(,"package")
[1] "edgeR"
[1] 27179 94 創建 DGEList 物件
首先簡化名稱
samplenames <- substring(colnames(x), 17, nchar(colnames(x)))
colnames(x) <- samplenames
samplenames[1] "10_6_5_11" "9_6_5_11" "purep53" "JMS8-2" "JMS8-3" "JMS8-4" "JMS8-5" "JMS9-P7c" "JMS9-P8c" 4.1 樣本資訊(samples)
Important
通常此步驟建議用 excel 或 csv 等檔案直接匯入樣本的 metadata
group <- as.factor(c("LP", "ML", "Basal", "Basal", "ML", "LP", "Basal", "ML", "LP"))
x$samples$group <- group
lane <- as.factor(rep(c("L004","L006","L008"), c(3,4,2)))
x$samples$lane <- lane
x$samples files group lib.size norm.factors lane
10_6_5_11 data/GSM1545535_10_6_5_11.txt LP 32863052 1 L004
9_6_5_11 data/GSM1545536_9_6_5_11.txt ML 35335491 1 L004
purep53 data/GSM1545538_purep53.txt Basal 57160817 1 L004
JMS8-2 data/GSM1545539_JMS8-2.txt Basal 51368625 1 L006
JMS8-3 data/GSM1545540_JMS8-3.txt ML 75795034 1 L006
JMS8-4 data/GSM1545541_JMS8-4.txt LP 60517657 1 L006
JMS8-5 data/GSM1545542_JMS8-5.txt Basal 55086324 1 L006
JMS9-P7c data/GSM1545544_JMS9-P7c.txt ML 21311068 1 L008
JMS9-P8c data/GSM1545545_JMS9-P8c.txt LP 19958838 1 L0084.2 準備基因對照(genes)
需要準備基因層級資訊,可以使用 biomaRt 調出所需資料
geneid <- rownames(x)
genes <- biomaRt::select(Mus.musculus, keys=geneid, columns=c("SYMBOL", "TXCHROM"),
keytype="ENTREZID")
head(genes)'select()' returned 1:many mapping between keys and columns
ENTREZID SYMBOL TXCHROM
1 497097 Xkr4 chr1
2 100503874 Gm19938 <NA>
3 100038431 Gm10568 <NA>
4 19888 Rp1 chr1
5 20671 Sox17 chr1
6 27395 Mrpl15 chr1
Important
必須確認是否有重複資訊,若有要處理,目前先簡化只取第一個!
genes <- genes[!duplicated(genes$ENTREZID),]
x$genes <- genes
xAn object of class "DGEList"
$samples
files group lib.size norm.factors lane
10_6_5_11 data/GSM1545535_10_6_5_11.txt LP 32863052 1 L004
9_6_5_11 data/GSM1545536_9_6_5_11.txt ML 35335491 1 L004
purep53 data/GSM1545538_purep53.txt Basal 57160817 1 L004
JMS8-2 data/GSM1545539_JMS8-2.txt Basal 51368625 1 L006
JMS8-3 data/GSM1545540_JMS8-3.txt ML 75795034 1 L006
JMS8-4 data/GSM1545541_JMS8-4.txt LP 60517657 1 L006
JMS8-5 data/GSM1545542_JMS8-5.txt Basal 55086324 1 L006
JMS9-P7c data/GSM1545544_JMS9-P7c.txt ML 21311068 1 L008
JMS9-P8c data/GSM1545545_JMS9-P8c.txt LP 19958838 1 L008
$counts
Samples
Tags 10_6_5_11 9_6_5_11 purep53 JMS8-2 JMS8-3 JMS8-4 JMS8-5 JMS9-P7c JMS9-P8c
497097 1 2 342 526 3 3 535 2 0
100503874 0 0 5 6 0 0 5 0 0
100038431 0 0 0 0 0 0 1 0 0
19888 0 1 0 0 17 2 0 1 0
20671 1 1 76 40 33 14 98 18 8
27174 more rows ...
$genes
ENTREZID SYMBOL TXCHROM
1 497097 Xkr4 chr1
2 100503874 Gm19938 <NA>
3 100038431 Gm10568 <NA>
4 19888 Rp1 chr1
5 20671 Sox17 chr1
27174 more rows ...
Important
以上是完整的 DGEList 物件 所需的資料,包含 samples, counts and genes
5 前處理
5.1 取 CPM
cpm <- cpm(x)
lcpm <- cpm(x, log=TRUE)L <- mean(x$samples$lib.size) * 1e-6
M <- median(x$samples$lib.size) * 1e-6
c(L, M)[1] 45.48855 51.36862summary(lcpm) 5535_10_6_5_11 5536_9_6_5_11 5538_purep53 5539_JMS8-2 5540_JMS8-3 5541_JMS8-4 5542_JMS8-5
Min. :-4.5074 Min. :-4.5074 Min. :-4.50743 Min. :-4.5074 Min. :-4.5074 Min. :-4.5074 Min. :-4.50743
1st Qu.:-4.5074 1st Qu.:-4.5074 1st Qu.:-4.50743 1st Qu.:-4.5074 1st Qu.:-4.5074 1st Qu.:-4.5074 1st Qu.:-4.50743
Median :-0.6847 Median :-0.3589 Median :-0.09513 Median :-0.0901 Median :-0.4281 Median :-0.4064 Median :-0.07152
Mean : 0.1714 Mean : 0.3312 Mean : 0.43559 Mean : 0.4089 Mean : 0.3225 Mean : 0.2529 Mean : 0.40428
3rd Qu.: 4.2913 3rd Qu.: 4.5601 3rd Qu.: 4.60081 3rd Qu.: 4.5475 3rd Qu.: 4.5772 3rd Qu.: 4.3199 3rd Qu.: 4.42513
Max. :14.7632 Max. :13.4952 Max. :12.95700 Max. :12.8513 Max. :12.9578 Max. :14.8520 Max. :13.19491
5544_JMS9-P7c 5545_JMS9-P8c
Min. :-4.5074 Min. :-4.5074
1st Qu.:-4.5074 1st Qu.:-4.5074
Median :-0.1704 Median :-0.3300
Mean : 0.3708 Mean : 0.2749
3rd Qu.: 4.6031 3rd Qu.: 4.4355
Max. :12.9413 Max. :14.0102 5.2 移除低表現基因
keep.exprs <- filterByExpr(x, group=group)
x <- x[keep.exprs,, keep.lib.sizes=FALSE]
dim(x)[1] 16624 95.3 繪圖
lcpm.cutoff <- log2(10/M + 2/L)
library(RColorBrewer)
nsamples <- ncol(x)
col <- brewer.pal(nsamples, "Paired")
png(filename = "./images/filter_cpm.png", width = 2000, height = 1000, res = 200)
par(mfrow=c(1,2))
plot(density(lcpm[,1]), col=col[1], lwd=2, ylim=c(0,0.26), las=2, main="", xlab="")
title(main="A. Raw data", xlab="Log-cpm")
abline(v=lcpm.cutoff, lty=3)
for (i in 2:nsamples){
den <- density(lcpm[,i])
lines(den$x, den$y, col=col[i], lwd=2)
}
legend("topright", samplenames, text.col=col, bty="n")
lcpm <- cpm(x, log=TRUE)
plot(density(lcpm[,1]), col=col[1], lwd=2, ylim=c(0,0.26), las=2, main="", xlab="")
title(main="B. Filtered data", xlab="Log-cpm")
abline(v=lcpm.cutoff, lty=3)
for (i in 2:nsamples){
den <- density(lcpm[,i])
lines(den$x, den$y, col=col[i], lwd=2)
}
legend("topright", samplenames, text.col=col, bty="n")
dev.off()
par(mfrow = c(1,1))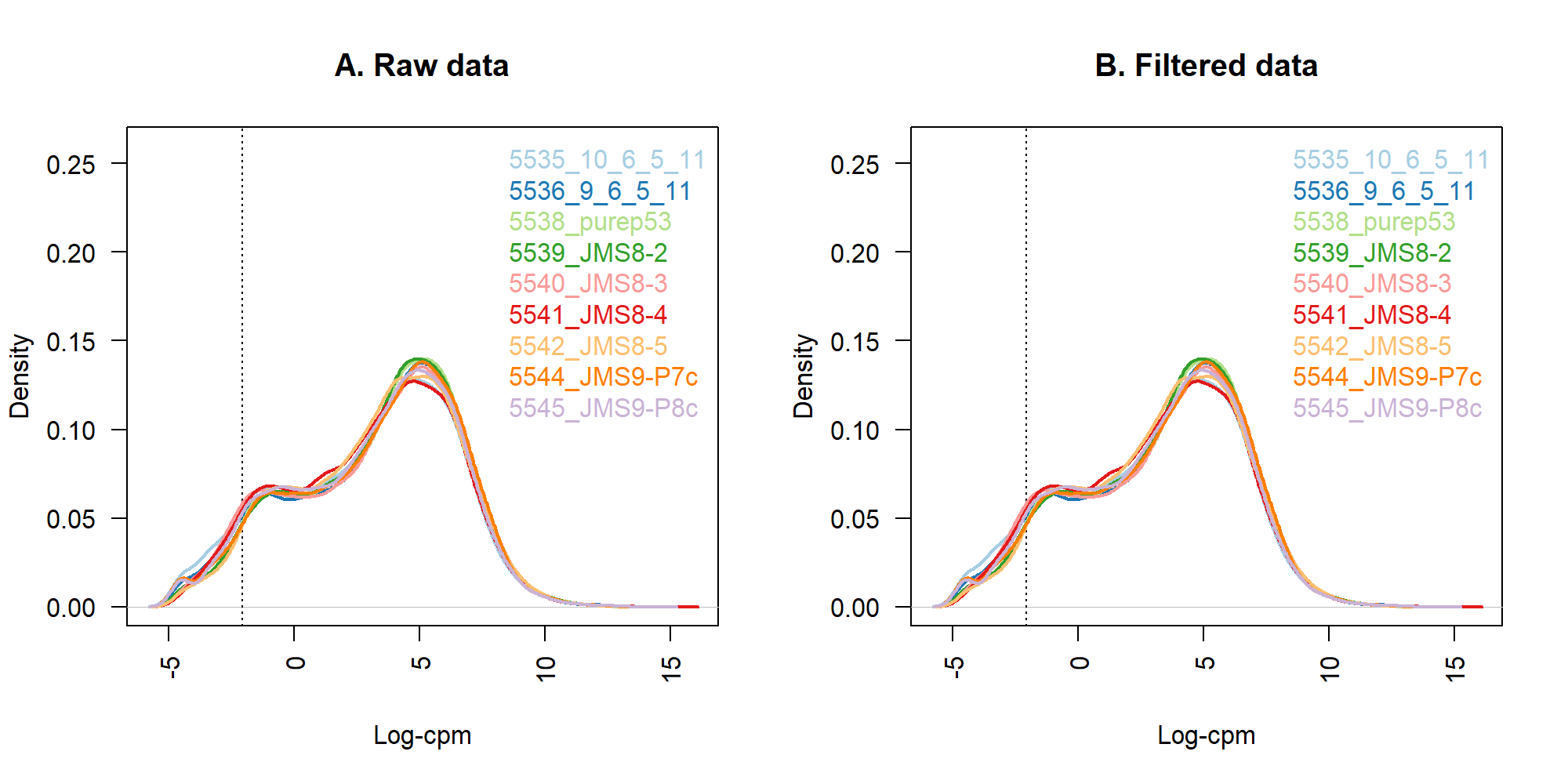
5.4 TMM 標準化
x <- calcNormFactors(x, method = "TMM")
x$samples$norm.factors# create toy data
x2 <- x
x2$samples$norm.factors <- 1
x2$counts[,1] <- ceiling(x2$counts[,1]*0.05)
x2$counts[,2] <- x2$counts[,2]*5
# plot setting
png(filename = "./images/tmm_boxplot.png", width = 2000, height = 1000, res = 200)
par(mfrow=c(1,2))
# boxplot before TMM
lcpm <- cpm(x2, log=TRUE)
boxplot(lcpm, las=2, col=col, main="")
title(main="A. Example: Unnormalised data",ylab="Log-cpm")
# perform TMM
x2 <- calcNormFactors(x2)
x2$samples$norm.factors
# boxplot after TMM
lcpm <- cpm(x2, log=TRUE)
boxplot(lcpm, las=2, col=col, main="")
title(main="B. Example: Normalised data",ylab="Log-cpm")
# plot setting
dev.off()
par(mfrow = c(1,1))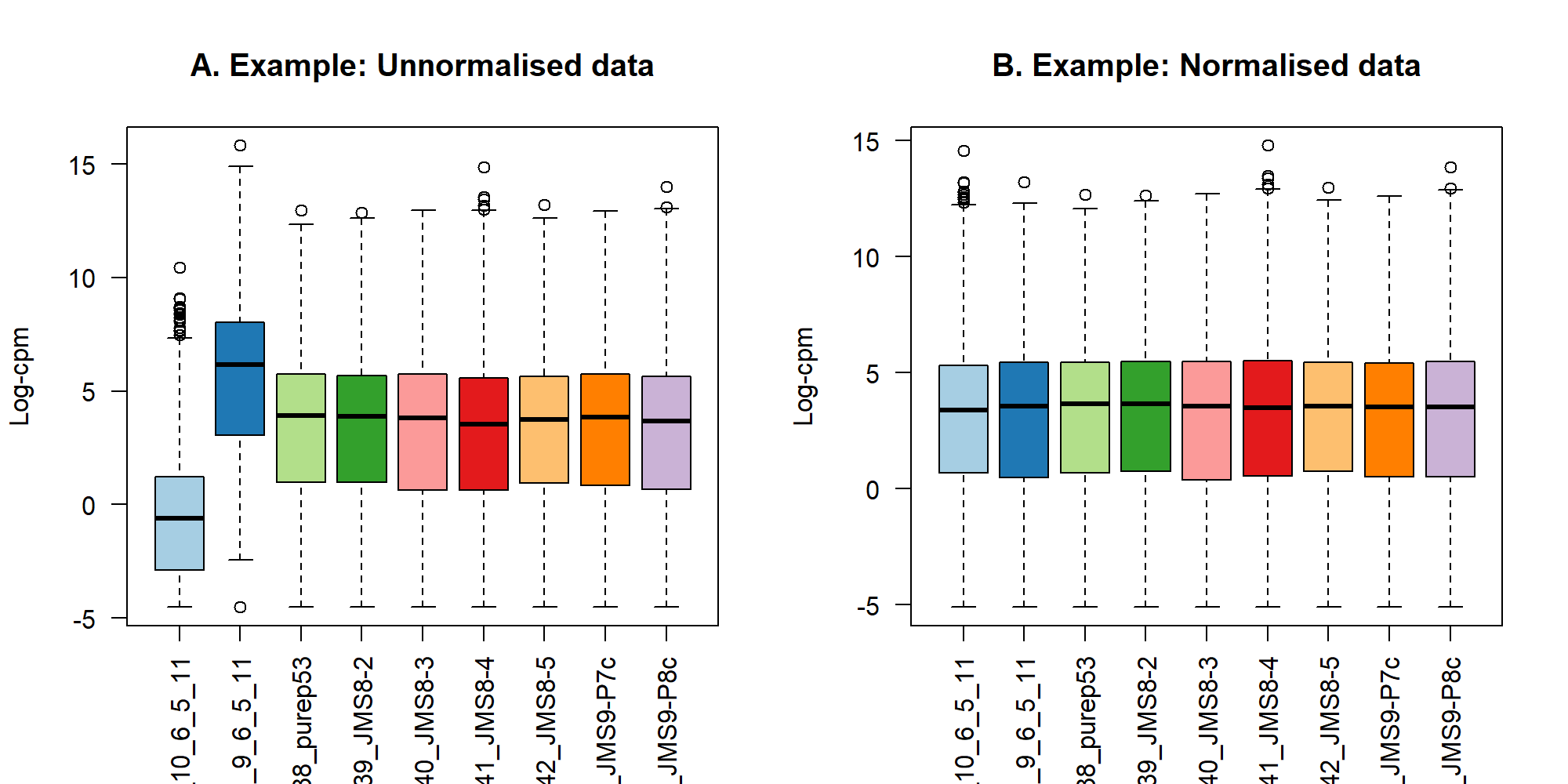
5.5 cluster 分析
png(filename = "./images/MDS.png", width = 2000, height = 1000, res = 200)
lcpm <- cpm(x, log=TRUE)
par(mfrow=c(1,2))
col.group <- group
levels(col.group) <- brewer.pal(nlevels(col.group), "Set1")
col.group <- as.character(col.group)
col.lane <- lane
levels(col.lane) <- brewer.pal(nlevels(col.lane), "Set2")
col.lane <- as.character(col.lane)
plotMDS(lcpm, labels=group, col=col.group)
title(main="A. Sample groups")
plotMDS(lcpm, labels=lane, col=col.lane, dim=c(3,4))
title(main="B. Sequencing lanes")
dev.off()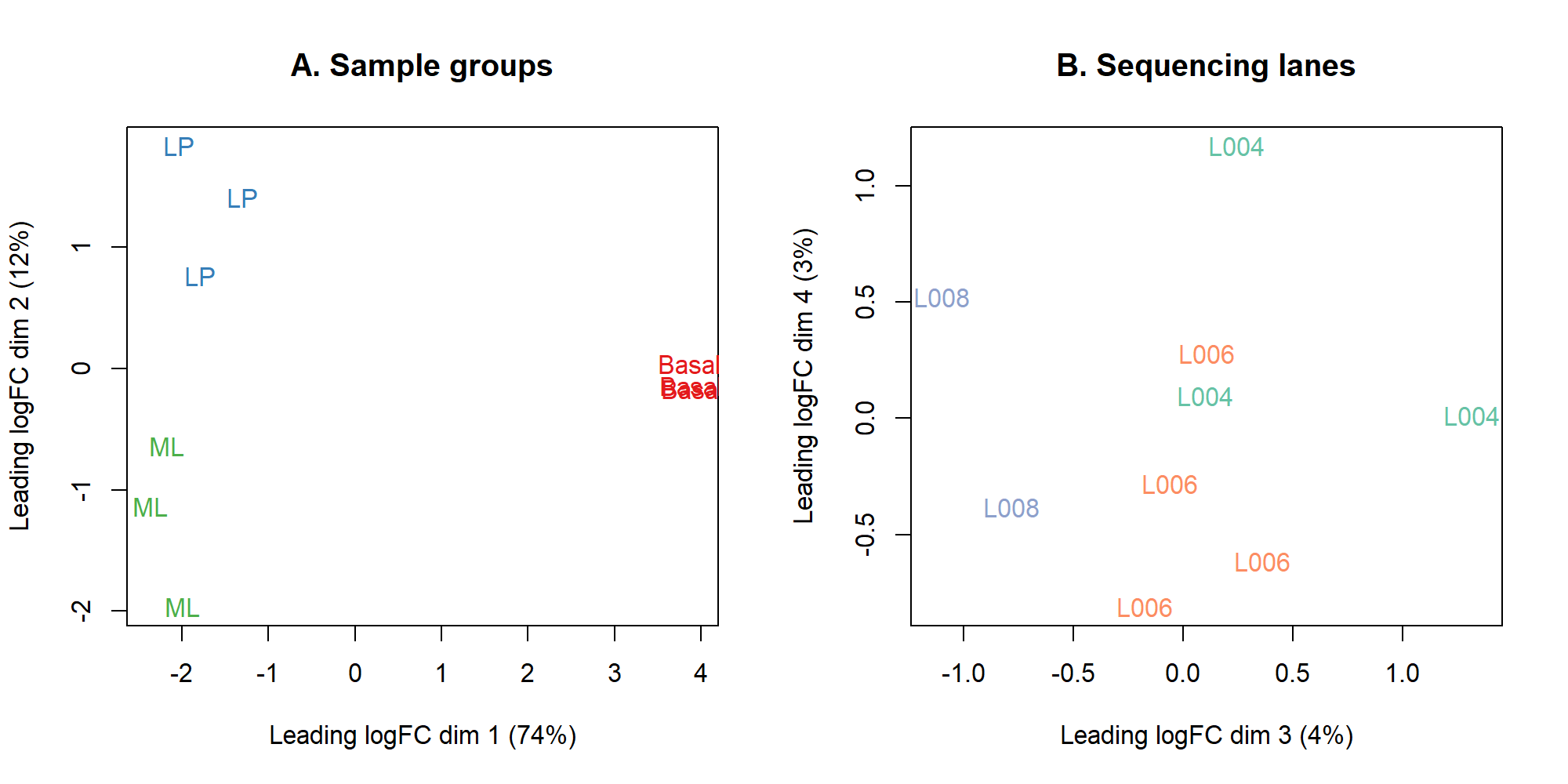
glMDSPlot(lcpm, labels=paste(group, lane, sep="_"),
groups=x$samples[,c(2,5)], launch=TRUE)6 DE 分析
6.1 建立 design 矩陣
design <- model.matrix(~0+group+lane)
colnames(design) <- gsub("group", "", colnames(design))
design Basal LP ML laneL006 laneL008
1 0 1 0 0 0
2 0 0 1 0 0
3 1 0 0 0 0
4 1 0 0 1 0
5 0 0 1 1 0
6 0 1 0 1 0
7 1 0 0 1 0
8 0 0 1 0 1
9 0 1 0 0 1
attr(,"assign")
[1] 1 1 1 2 2
attr(,"contrasts")
attr(,"contrasts")$group
[1] "contr.treatment"
attr(,"contrasts")$lane
[1] "contr.treatment"6.2 建立比較矩陣
contr.matrix <- makeContrasts(
BasalvsLP = Basal-LP,
BasalvsML = Basal - ML,
LPvsML = LP - ML,
levels = colnames(design))
contr.matrix Contrasts
Levels BasalvsLP BasalvsML LPvsML
Basal 1 1 0
LP -1 0 1
ML 0 -1 -1
laneL006 0 0 0
laneL008 0 0 06.3 voom
png(filename = "./images/voom.png", width = 2000, height = 1000, res = 200)
par(mfrow=c(1,2))
v <- voom(x, design, plot=TRUE)
v
vfit <- lmFit(v, design)
vfit <- contrasts.fit(vfit, contrasts=contr.matrix)
efit <- eBayes(vfit)
plotSA(efit, main="Final model: Mean-variance trend")
dev.off()
par(mfrow=c(1,1))An object of class "EList"
$genes
ENTREZID SYMBOL TXCHROM
1 497097 Xkr4 chr1
5 20671 Sox17 chr1
6 27395 Mrpl15 chr1
7 18777 Lypla1 chr1
9 21399 Tcea1 chr1
16619 more rows ...
$targets
files group lib.size norm.factors lane
5535_10_6_5_11 data/GSM1545535_10_6_5_11.txt LP 29387429 0.8943956 L004
5536_9_6_5_11 data/GSM1545536_9_6_5_11.txt ML 36212498 1.0250186 L004
5538_purep53 data/GSM1545538_purep53.txt Basal 59771061 1.0459005 L004
5539_JMS8-2 data/GSM1545539_JMS8-2.txt Basal 53711278 1.0458455 L006
5540_JMS8-3 data/GSM1545540_JMS8-3.txt ML 77015912 1.0162707 L006
5541_JMS8-4 data/GSM1545541_JMS8-4.txt LP 55769890 0.9217132 L006
5542_JMS8-5 data/GSM1545542_JMS8-5.txt Basal 54863512 0.9961959 L006
5544_JMS9-P7c data/GSM1545544_JMS9-P7c.txt ML 23139691 1.0861026 L008
5545_JMS9-P8c data/GSM1545545_JMS9-P8c.txt LP 19634459 0.9839203 L008
$E
Samples
Tags 5535_10_6_5_11 5536_9_6_5_11 5538_purep53 5539_JMS8-2 5540_JMS8-3 5541_JMS8-4 5542_JMS8-5 5544_JMS9-P7c 5545_JMS9-P8c
497097 -4.292165 -3.856488 2.5185849 3.2931366 -4.459730 -3.994060 3.2869677 -3.2103696 -5.295316
20671 -4.292165 -4.593453 0.3560126 -0.4073032 -1.200995 -1.943434 0.8442767 -0.3228444 -1.207853
27395 3.876089 4.413107 4.5170045 4.5617546 4.344401 3.786363 3.8990635 4.3396075 4.124644
18777 4.708774 5.571872 5.3964008 5.1623650 5.649355 5.081611 5.0602470 5.7513694 5.142436
21399 4.785541 4.754537 5.3703795 5.1220551 4.869586 4.943840 5.1384776 5.0308985 4.979644
16619 more rows ...
$weights
[,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9]
[1,] 1.079413 1.332986 19.826915 20.273253 1.993686 1.395853 20.494977 1.107780 1.079413
[2,] 1.170357 1.456380 4.804866 8.660025 3.612508 2.626870 8.760149 3.211473 2.541942
[3,] 20.219073 25.573792 30.434759 28.528310 31.352260 25.743247 28.722497 21.200072 16.657930
[4,] 26.947557 32.505933 33.583128 33.232125 34.231754 32.354158 33.334340 30.348630 24.259801
[5,] 26.610864 28.501638 33.645479 33.206374 33.573492 31.996623 33.308490 25.171513 23.573305
16619 more rows ...
$design
Basal LP ML laneL006 laneL008
1 0 1 0 0 0
2 0 0 1 0 0
3 1 0 0 0 0
4 1 0 0 1 0
5 0 0 1 1 0
6 0 1 0 1 0
7 1 0 0 1 0
8 0 0 1 0 1
9 0 1 0 0 1
attr(,"assign")
[1] 1 1 1 2 2
attr(,"contrasts")
attr(,"contrasts")$group
[1] "contr.treatment"
attr(,"contrasts")$lane
[1] "contr.treatment"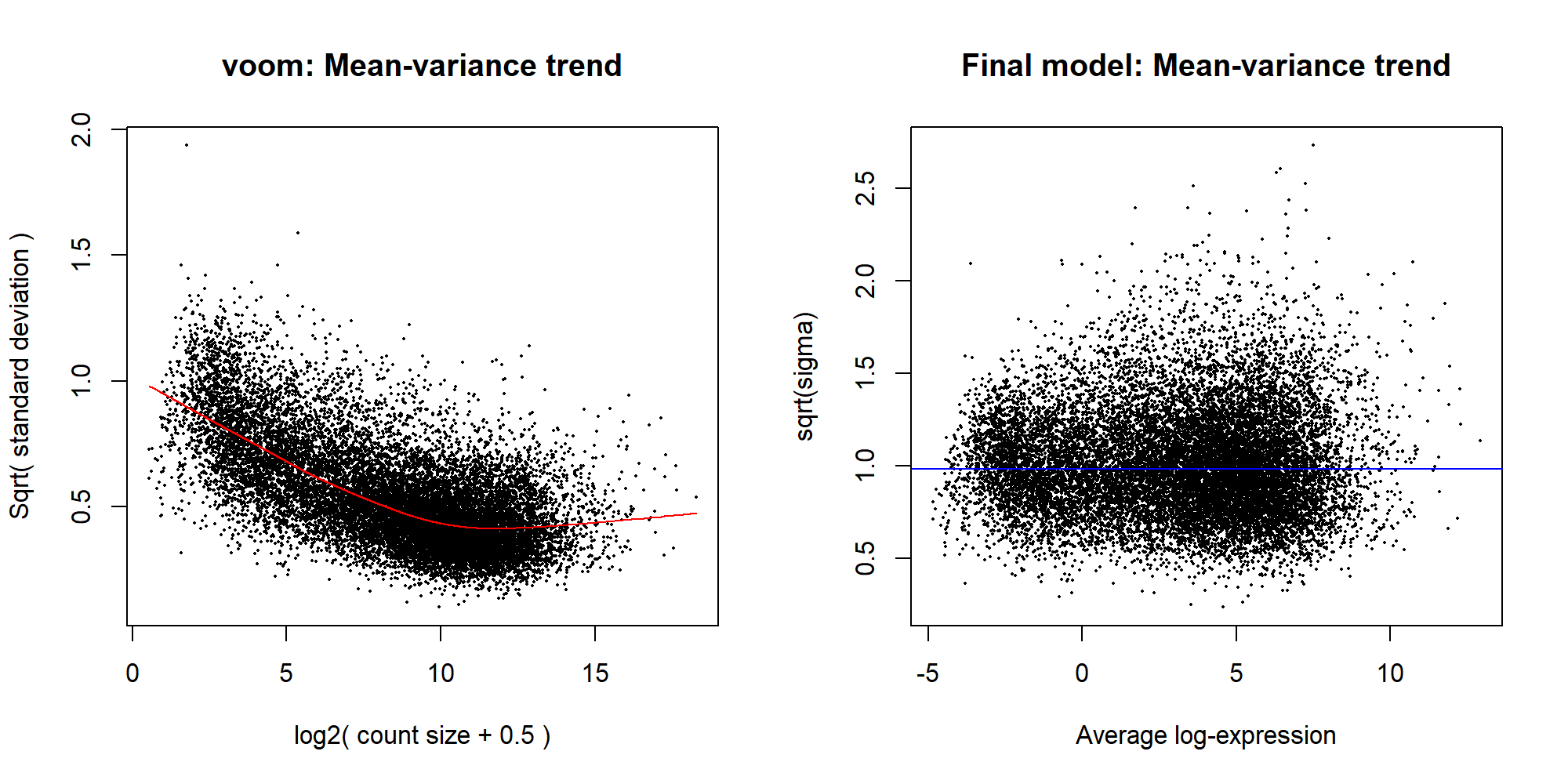
6.4 結果
summary(decideTests(efit)) BasalvsLP BasalvsML LPvsML
Down 4648 4927 3135
NotSig 7113 7026 10972
Up 4863 4671 2517tfit <- treat(vfit, lfc=1)
dt <- decideTests(tfit)
summary(dt) BasalvsLP BasalvsML LPvsML
Down 1632 1777 224
NotSig 12976 12790 16210
Up 2016 2057 190de.common <- which(dt[,1]!=0 & dt[,2]!=0)
length(de.common)
head(tfit$genes$SYMBOL[de.common], n=20)
png(filename = "./images/venn.png", width = 1500, height = 1200, res = 200)
vennDiagram(dt[,1:2], circle.col=c("turquoise", "salmon"))
dev.off()
write.fit(tfit, dt, file="results/results.txt")[1] 2784
[1] "Xkr4" "Rgs20" "Cpa6" "A830018L16Rik" "Sulf1" "Eya1" "Msc" "Sbspon"
[9] "Pi15" "Crispld1" "Kcnq5" "Rims1" "Khdrbs2" "Ptpn18" "Prss39" "Arhgef4"
[17] "Cnga3" "Cracdl" "Aff3" "Npas2" 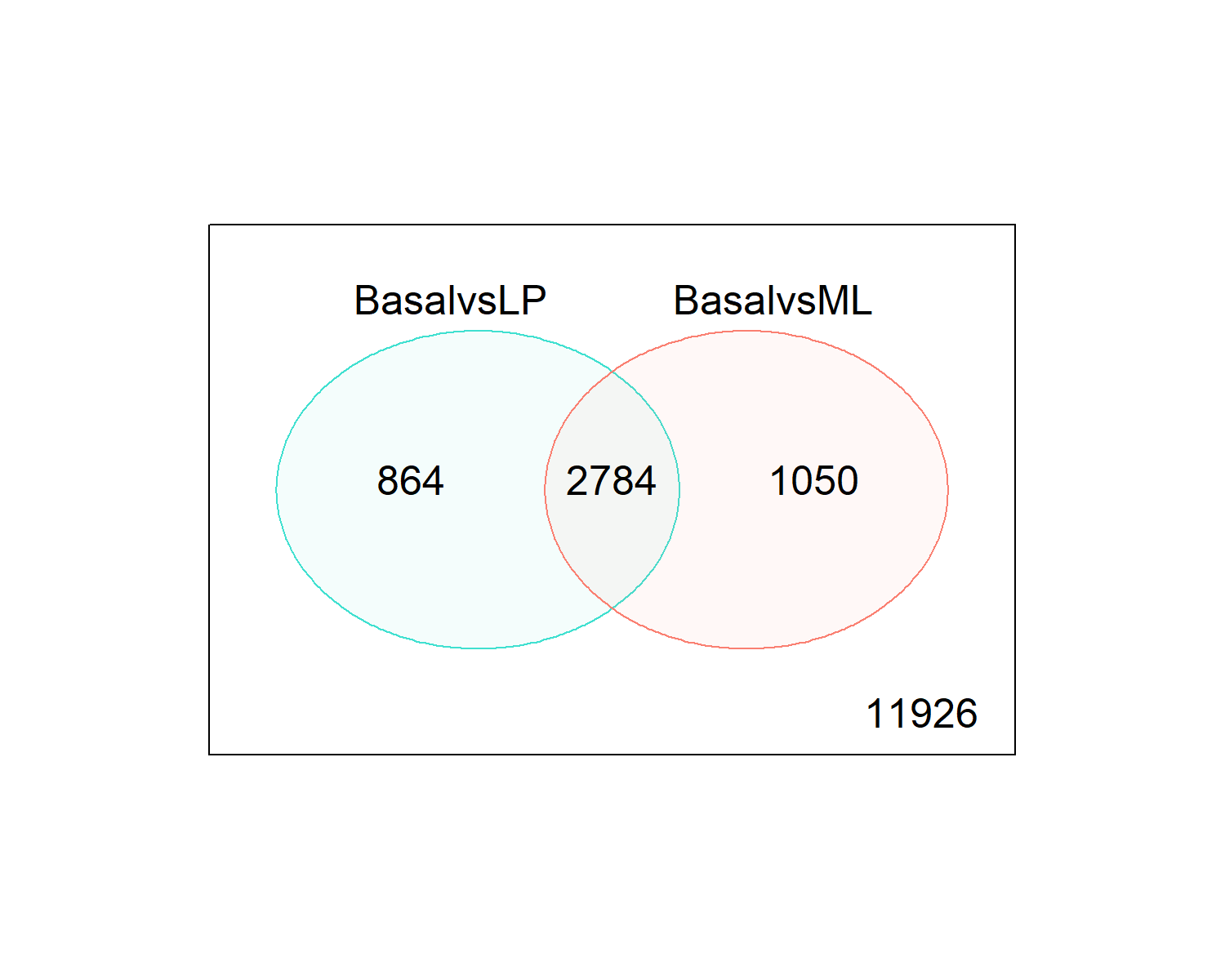
basal.vs.lp <- topTreat(tfit, coef=1, n=Inf)
basal.vs.ml <- topTreat(tfit, coef=2, n=Inf)
head(basal.vs.lp) ENTREZID SYMBOL TXCHROM logFC AveExpr t P.Value adj.P.Val
12759 12759 Clu chr14 -5.455444 8.856581 -33.55508 1.723731e-10 1.707586e-06
53624 53624 Cldn7 chr11 -5.527356 6.295437 -31.97515 2.576972e-10 1.707586e-06
242505 242505 Rasef chr4 -5.935249 5.118259 -31.33407 3.081544e-10 1.707586e-06
67451 67451 Pkp2 chr16 -5.738665 4.419170 -29.85616 4.575686e-10 1.739242e-06
228543 228543 Rhov chr2 -6.264208 5.485314 -29.07484 5.782844e-10 1.739242e-06
70350 70350 Basp1 chr15 -6.084738 5.247023 -28.26649 7.267694e-10 1.739242e-066.5 MA plot
png("images/MA-plot.png")
plotMD(tfit, column=1, status=dt[,1], main=colnames(tfit)[1],
xlim=c(-8,13))
dev.off()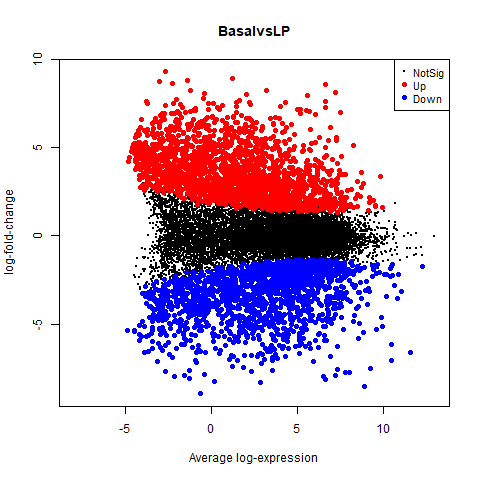
glMDPlot(tfit, coef=1, status=dt, main=colnames(tfit)[1],
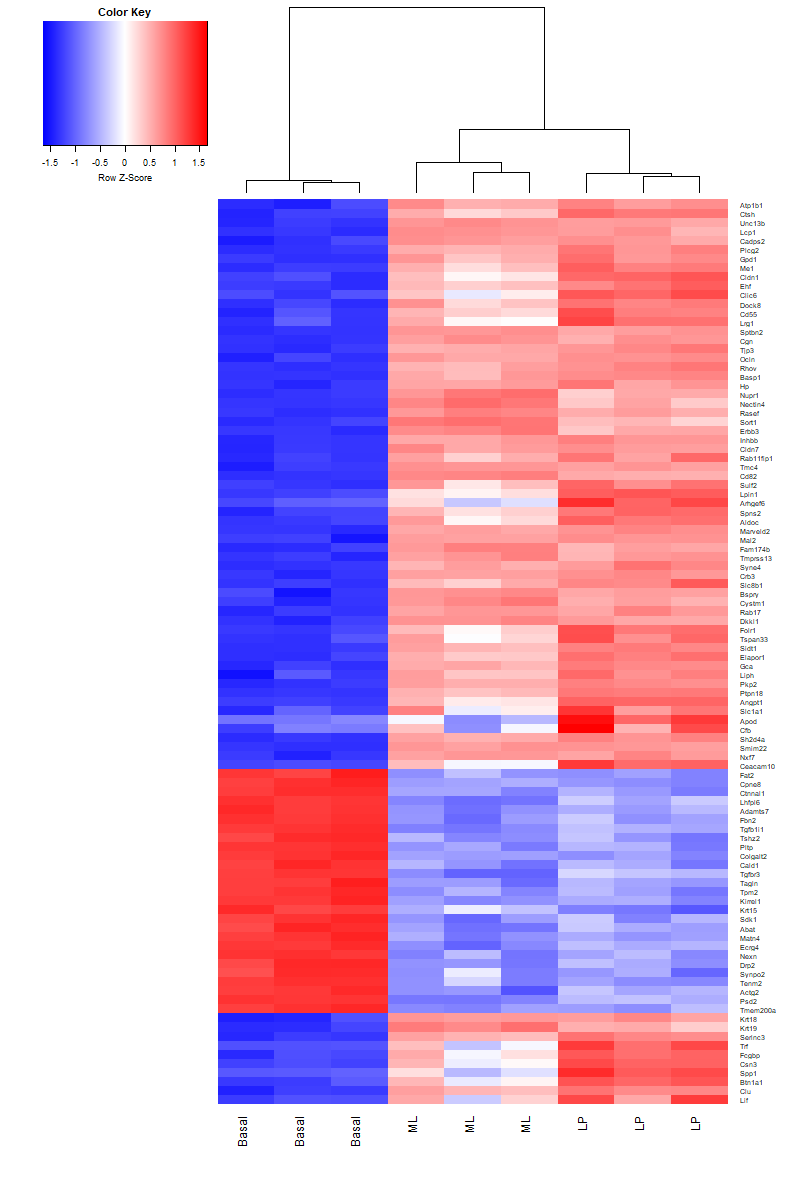
side.main="ENTREZID", counts=lcpm, groups=group, launch=TRUE)6.6 heatmap
library(gplots)
basal.vs.lp.topgenes <- basal.vs.lp$ENTREZID[1:100]
i <- which(v$genes$ENTREZID %in% basal.vs.lp.topgenes)
mycol <- colorpanel(1000,"blue","white","red")
png("images/heatmap.png", width = 800 ,height = 1200)
heatmap.2(lcpm[i,], scale="row",
labRow=v$genes$SYMBOL[i], labCol=group,
col=mycol, trace="none", density.info="none",
margin=c(8,6), lhei=c(2,10), dendrogram="column")
dev.off()