flowchart TD A[Multi-omics data] --> B[Design matrix] B --> C[Initial DIABLO model] C --> D[Cross validation] D --> E[Select components] E --> F[Tune feature number] F --> G[Final DIABLO model] G --> H[Visualization] G --> I[Model evaluation] I --> J[External validation]
multiomics-diablo 基本介紹
1 分析流程
2 DIABLO 介紹
在多體學(multi-omics)研究中,常會同時取得不同層級的生物資料,例如 mRNA、miRNA、proteomics 或 metabolomics。這些資料反映不同的分子調控機制,但若分開分析,往往難以完整理解其系統性關聯。因此,多體學整合方法被廣泛用於同時分析多個 omics 資料,以探索跨層級的生物訊號與潛在 biomarker。
DIABLO(Data Integration Analysis for Biomarker discovery using Latent variable approaches for Omics studies) 是 mixOmics 套件提供的一種監督式多體學整合方法。其核心基於 block sparse Partial Least Squares Discriminant Analysis (block sPLS-DA),透過建立 latent components,同時最大化不同 omics 資料之間的相關性與 phenotype 的分類能力。DIABLO 也會透過稀疏特徵選擇(sparse feature selection),挑選少量但具有代表性的分子特徵,以建立可解釋的多體學 biomarker signature。
ImportantDIABLO vs. MINT
| 方法 | 解決問題 | 整合什麼 |
|---|---|---|
| MINT | multi-study integration | 不同研究資料 |
| DIABLO | multi-omics integration | 不同 omics 資料 |
Tiplatent component 是什麼?
latent component = 多個原始特徵加權組合形成的一個新變數
在本範例中,我們使用 mixOmics 套件內建的
TCGA breast cancer multi-omics dataset,整合 mRNA、miRNA 與 proteomics 三種資料,示範 DIABLO 的基本分析流程,包括模型建立、參數 tuning、特徵選擇、結果視覺化以及模型評估。透過此流程,可以同時探索不同 omics 層級之間的關聯,並建立具有預測能力的多體學分類模型。
TipReference
This tutorial is a rewritten Quarto version of the DIABLO case study originally provided by the mixOmics team.
Original tutorial: - mixomics.org
- Vignette - mixOmicsTeam/mixOmics github
Reference: Rohart F., Gautier, B, Singh, A and Lê Cao, K. A. mixOmics: an R package for ’omics feature selection and multiple data integration. PLOS 2-17.
3 下載套件
if (!requireNamespace("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
if (!requireNamespace("mixOmics", quietly = TRUE)) {
BiocManager::install("mixOmics")
}
library(mixOmics)
Noteoutputs
Loading required package: MASS
Loading required package: lattice
Loading required package: ggplot2
Need help? Try Stackoverflow: https://stackoverflow.com/tags/ggplot2
Loaded mixOmics 6.30.0
Thank you for using mixOmics!
Tutorials: http://mixomics.org
Bookdown vignette: https://mixomicsteam.github.io/Bookdown
Questions, issues: Follow the prompts at http://mixomics.org/contact-us
Cite us: citation('mixOmics')4 範例資料
使用 mixOmics 套件內建的 breast.TCGA 資料集作為範例,其中有 mRNA, miRNA 與 proteomics 三種體學資料的 matrix,將其整合成一個 list
data("breast.TCGA")
set.seed(123)
data <- list(
mRNA = breast.TCGA$data.train$mrna,
miRNA = breast.TCGA$data.train$mirna,
proteomics = breast.TCGA$data.train$protein
)
lapply(data, dim)$mRNA
[1] 150 200
$miRNA
[1] 150 184
$proteomics
[1] 150 142Y 表示每個樣本的乳癌分型(Basal、Her2、LumA),作為 DIABLO 模型的分類標籤(response variable)。
Y <- breast.TCGA$data.train$subtype
summary(Y)Basal Her2 LumA
45 30 75 5 設計 design matrix
在 DIABLO 分析中,design matrix 用來定義不同 omics 資料之間的關聯程度。矩陣中的數值介於 0 到 1 之間,數值越大代表模型在建立 latent components 時,越強調不同資料區塊之間的相關性。
- 0:表示不強制建立跨體學關聯
- 接近 1:表示強化不同 omics 之間的相關性
- 0.1–0.5:常作為中等程度的整合強度
在此範例中,三個 omics 資料(mRNA、miRNA、proteomics)之間設定為 0.1 的弱關聯,表示模型會考慮跨體學的相關性,但不會過度強制不同資料之間必須高度一致。對角線設為 0,因為每個資料區塊本身不需要與自己建立關聯。
design <- matrix(
0.1,
ncol = length(data),
nrow = length(data),
dimnames = list(names(data), names(data))
)
diag(design) <- 0
design mRNA miRNA proteomics
mRNA 0.0 0.1 0.1
miRNA 0.1 0.0 0.1
proteomics 0.1 0.1 0.06 初始 DIABLO 模型
使用 block.splsda() 建立 DIABLO 模型。此方法為 mixOmics 中用於多體學整合的監督式學習模型,可同時整合多個 omics 資料區塊並建立分類模型。 在此設定 ncomp = 5,表示模型最多建立 5 個 latent components,用於捕捉不同 omics 資料中的共同變異結構與分類訊號。
模型建立後,使用 perf() 進行交叉驗證(cross-validation)以評估模型效能。此範例採用 10-fold cross-validation 並重複 10 次,以估計模型在不同 component 數量下的分類錯誤率,並協助選擇最佳的 component 數量。
此外,利用 proc.time() 記錄模型評估所需的運算時間。
sgccda.res <- block.splsda(
X = data,
Y = Y,
ncomp = 5,
design = design
)
t1 <- proc.time()
perf.diablo <- perf(
sgccda.res,
validation = "Mfold",
folds = 10,
nrepeat = 10
)
running_time <- proc.time() - t1
running_timeDesign matrix has changed to include Y; each block will be
linked to Y.
user system elapsed
18.03 0.60 18.77 6.1 視覺化結果
plot(perf.diablo) 會顯示在不同 component 數量下模型的分類錯誤率,用來評估模型效能並協助選擇適合的 component 數量。
圖中會呈現不同距離計算方法(例如 max.dist、centroids.dist、mahalanobis.dist)所對應的錯誤率變化。隨著 component 數量增加,錯誤率通常會先下降後趨於穩定,因此可以依據最低錯誤率的位置來決定最佳的 component 數量。
png(filename = "./images/perf.png", width = 1500, height = 1500, res = 200)
plot(perf.diablo)
dev.off()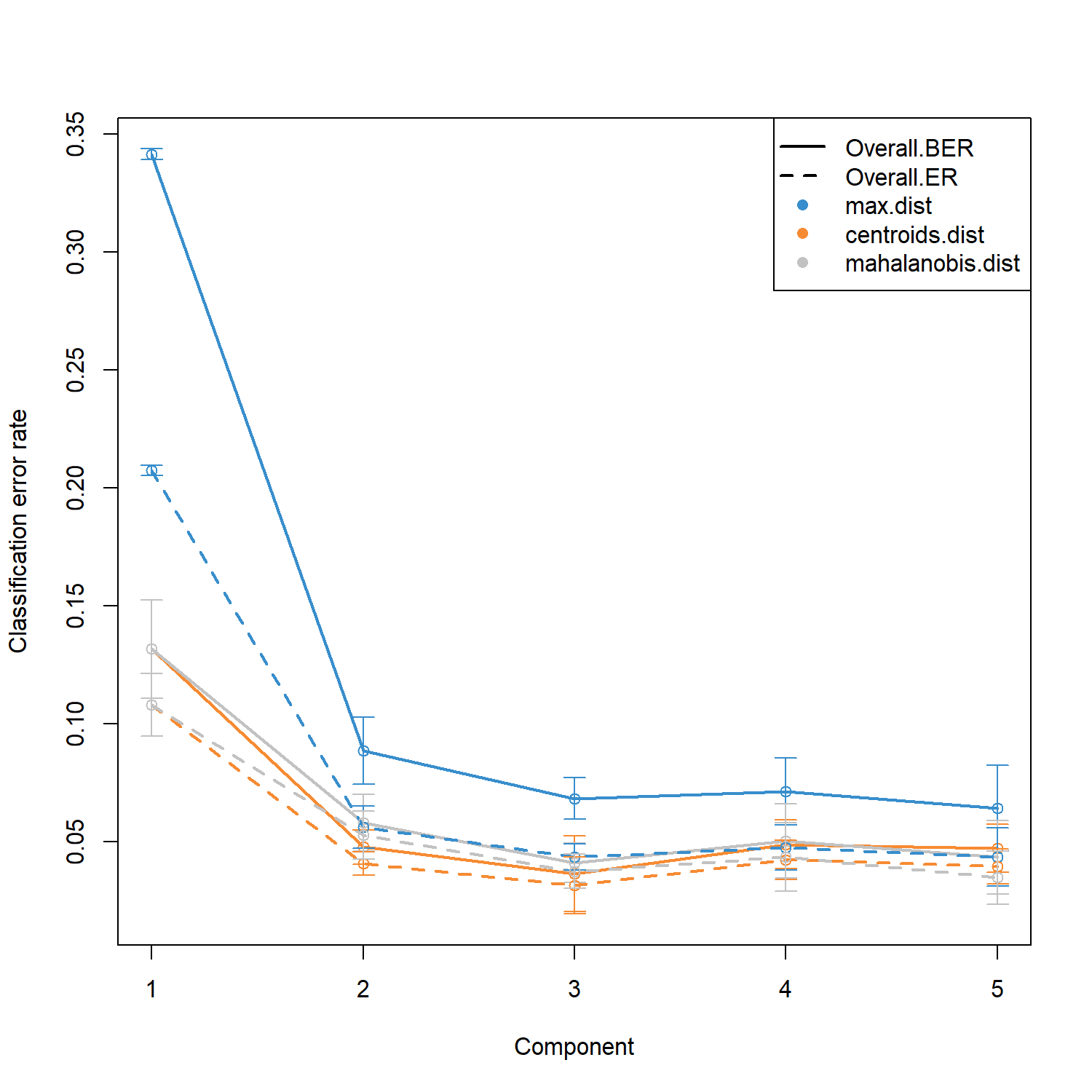
7 最佳 component 數量
choice.ncomp 會依據不同距離計算方式與錯誤率指標,回傳最適合的 component 數量。
其中 WeightedVote 為 DIABLO 分類模型常用的投票方式,會整合不同 omics 資料區塊的預測結果,以加權方式決定最終分類結果。
perf.diablo$choice.ncomp$WeightedVote max.dist centroids.dist mahalanobis.dist
Overall.ER 3 2 3
Overall.BER 3 2 3根據 perf() 的結果,接著選擇最適合的 component 數量 (ncomp)。此處使用 Balanced Error Rate (BER) 作為主要評估指標,並採用 centroids.dist 作為分類距離方法。
BER 可以在類別數量不平衡的情況下提供較公平的分類評估,因此在多分類問題中常被用來作為模型選擇的依據。
ncomp <- perf.diablo$choice.ncomp$WeightedVote["Overall.BER", "centroids.dist"]
ncomp[1] 28 特徵數量調整
在確定最佳的 component 數量後,接著需要決定每個 omics 資料區塊應保留多少特徵(keepX)。DIABLO 採用稀疏模型(sparse model),會從高維資料中選擇少量最具代表性的分子特徵,因此需要透過 tune.block.splsda() 來尋找最佳的特徵數量。
Important如果你的系統是 Windows,請跳過此步驟。
為了加速計算,本範例使用 BiocParallel 套件進行 平行運算,並設定 4 個 CPU workers。同時設定 test.keepX,定義每個 omics 資料區塊在不同 component 下要測試的特徵數量範圍。
if (!requireNamespace("BiocParallel", quietly = TRUE)) {
BiocManager::install("BiocParallel")
}
library(BiocParallel)
BPPARAM <- MulticoreParam(workers = 4)接著利用 tune.block.splsda() 進行參數調整,透過 10-fold cross-validation 評估不同 keepX 組合的分類表現,並找出最適合的特徵數量。
test.keepX <- list(
mRNA = c(5:9, seq(10, 18, 2), seq(20, 30, 5)),
miRNA = c(5:9, seq(10, 18, 2), seq(20, 30, 5)),
proteomics = c(5:9, seq(10, 18, 2), seq(20, 30, 5))
)
t1 <- proc.time()
tune.TCGA <- tune.block.splsda(
X = data,
Y = Y,
ncomp = ncomp,
test.keepX = test.keepX,
design = design,
validation = "Mfold",
folds = 10,
nrepeat = 1,
BPPARAM = BPPARAM,
dist = "centroids.dist"
)Design matrix has changed to include Y; each block will be
linked to Y.
You have provided a sequence of keepX of length: 13 for block mRNA and 13 for block miRNA and 13 for block proteomics.
This results in 2197 models being fitted for each component and each nrepeat, this may take some time to run, be patient!完成 tune.block.splsda() 之後,可以從結果中取得每個 omics 資料區塊在不同 component 下的 最佳特徵數量 (keepX)。這些數值代表在模型中保留的變數數量,也就是最終被選出的潛在 biomarker 數量。
list.keepX <- tune.TCGA$choice.keepX
tune.TCGA$choice.keepX$mRNA
[1] 30 7
$miRNA
[1] 9 16
$proteomics
[1] 7 59 最終 DIABLO 模型
在完成 component 與特徵數量 (keepX) 的調整後,接著建立最終 DIABLO 模型。此模型會使用先前選出的最佳 ncomp 與 keepX 參數,以整合不同 omics 資料並建立分類模型。
sgccda.res <- block.splsda(
X = data,
Y = Y,
ncomp = ncomp,
keepX = list.keepX,
design = design
)Design matrix has changed to include Y; each block will be
linked to Y.可以檢視模型所使用的 design matrix,確認不同 omics 資料區塊之間的整合關係。
sgccda.res$design mRNA miRNA proteomics Y
mRNA 0.0 0.1 0.1 1
miRNA 0.1 0.0 0.1 1
proteomics 0.1 0.1 0.0 1
Y 1.0 1.0 1.0 0DIABLO 也會在每個 component 中選出具有代表性的特徵。使用 selectVar() 可以取得模型所選出的變數,例如以下程式碼顯示 mRNA block 在 component 1 所選出的基因名稱。
selectVar(sgccda.res, block = 'mRNA', comp = 1)$mRNA$name [1] "ZNF552" "KDM4B" "CCNA2" "LRIG1" "PREX1" "FUT8" "C4orf34" "TTC39A" "ASPM" "SLC43A3" "MEX3A" "SEMA3C"
[13] "E2F1" "STC2" "FMNL2" "LMO4" "MED13L" "DTWD2" "CSRP2" "NTN4" "KIF13B" "SLC19A2" "NCAPG2" "FAM63A"
[25] "EPHB3" "MEGF9" "MTL5" "HTRA1" "SLC5A6" "SNORA8" 這些被選出的特徵即為 DIABLO 模型所識別的 潛在 multi-omics biomarker,可用於後續的生物學解釋或功能分析。
10 視覺化
10.1 plotDIABLO
plotDiablo() 可用來觀察不同 omics 資料區塊在 latent component 空間中的整合情形,並顯示不同資料區塊之間的 correlation。
Important目的
評估不同 omics block 所建立的 latent components 是否捕捉到共同的 biological signal。
Tip如何解讀 plotDiablo 圖
plotDiablo() 會以 pairwise matrix 的方式顯示不同 omics block 的 component 關係:
1️⃣ 對角線 (diagonal)
顯示各 omics block 的名稱,例如 mRNA、miRNA、proteomics。
2️⃣ 右上角 (upper triangle)
為 scatter plot,表示兩個 omics block 在指定 component(例如 component 1)上的樣本分布關係。
每個點代表一個樣本,顏色表示不同分類(例如 Basal、Her2、LumA)。
若不同 omics 的 component 捕捉到相似的 biological signal,樣本分布方向通常會呈現一致的趨勢。
3️⃣ 左下角 (lower triangle)
顯示兩個 omics block component 之間的 Pearson correlation coefficient。
數值越高代表不同 omics 在該 component 上的訊號越一致。
一般而言:
correlation > 0.7:表示不同 omics 在該 component 上具有良好的整合效果
correlation 0.4–0.7:中等相關
< 0.4:不同 omics 捕捉到的訊號可能較不一致
因此,plotDiablo() 主要用於評估 DIABLO 是否成功在不同 omics 中捕捉到共同的 biological signal。
png(filename = "./images/diablo.png", width = 1500, height = 1500, res = 200)
plotDiablo(sgccda.res, ncomp = 1)
dev.off()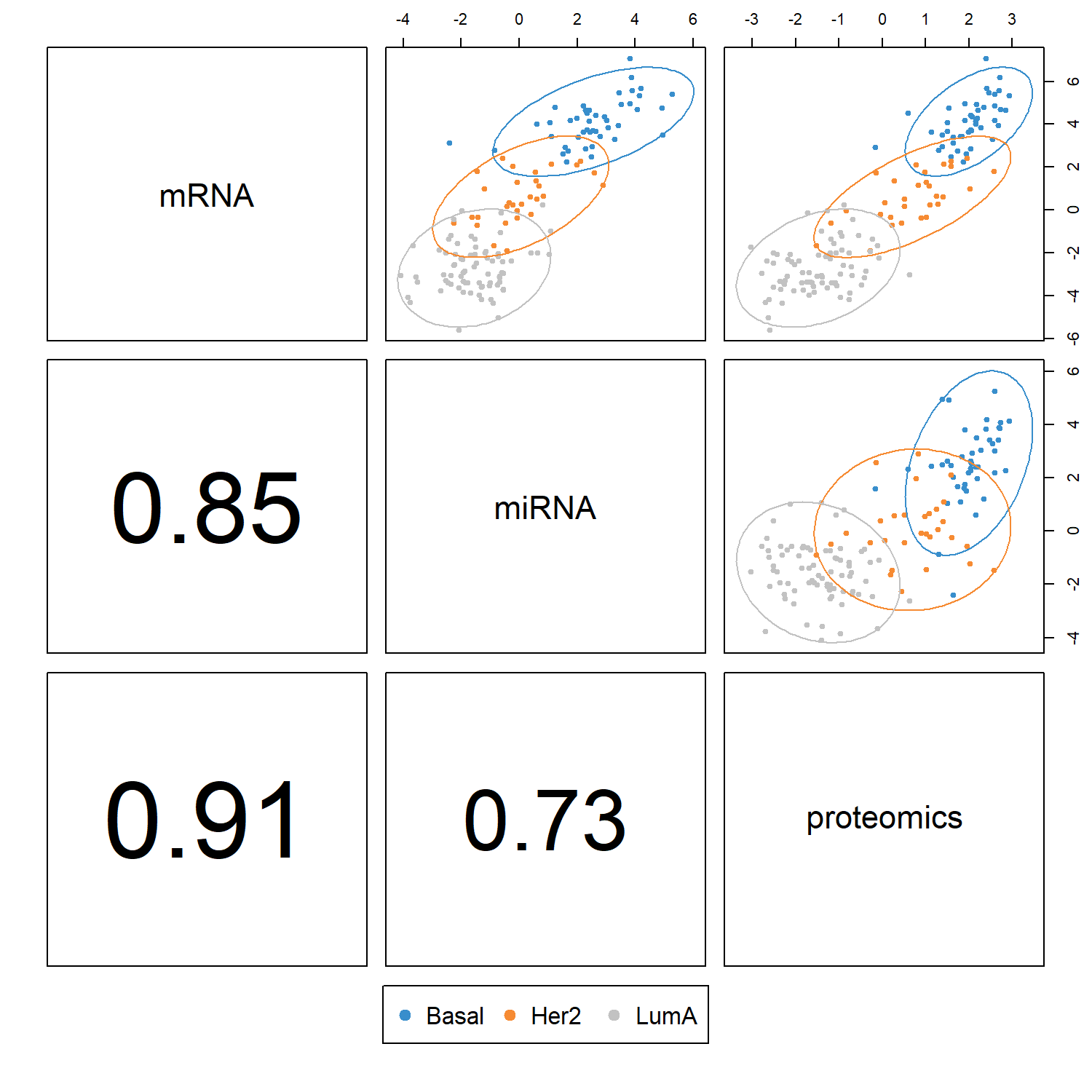
10.2 plotIndiv
plotIndiv() 用於顯示 樣本 在 component 空間中 的分布情況,可觀察不同分類(如癌症 subtype)是否能夠被清楚區分。
Important目的
觀察樣本在 latent component 空間中的分布情形,並評估模型是否能有效區分不同的 phenotype(例如癌症 subtype)。
Tip如何解讀 plotIndiv 圖
plotIndiv() 會將每個樣本投影到 DIABLO 所建立的 component 空間 中:
1️⃣ 每個點代表一個樣本 (sample)
每個點的位置由該樣本在 component 空間中的 score 決定。
2️⃣ 顏色代表不同分類 (group / phenotype)
例如本範例中的 Basal、Her2、LumA。
3️⃣ 座標軸代表 latent components
例如:
Component 1Component 2
這些 component 是由多個 omics feature 的加權組合所形成的潛在變數。
4️⃣ 群集分離程度 (cluster separation)
若不同分類的樣本在圖中形成清楚分離的群集,表示:
- 模型成功捕捉到與 phenotype 相關的 biological signal
- 不同分類在多體學資料中具有可辨識的分子特徵
若群集之間高度重疊,則可能表示:
- 分類訊號較弱
- component 數量不足
- 或模型需要重新調整參數。
因此,plotIndiv() 主要用於 評估 DIABLO 模型在樣本層級的分類能力與群集分離效果。
png(filename = "./images/indiv.png", width = 1500, height = 1500, res = 200)
plotIndiv(sgccda.res, ind.names = FALSE, legend = TRUE, style="ggplot2")
dev.off()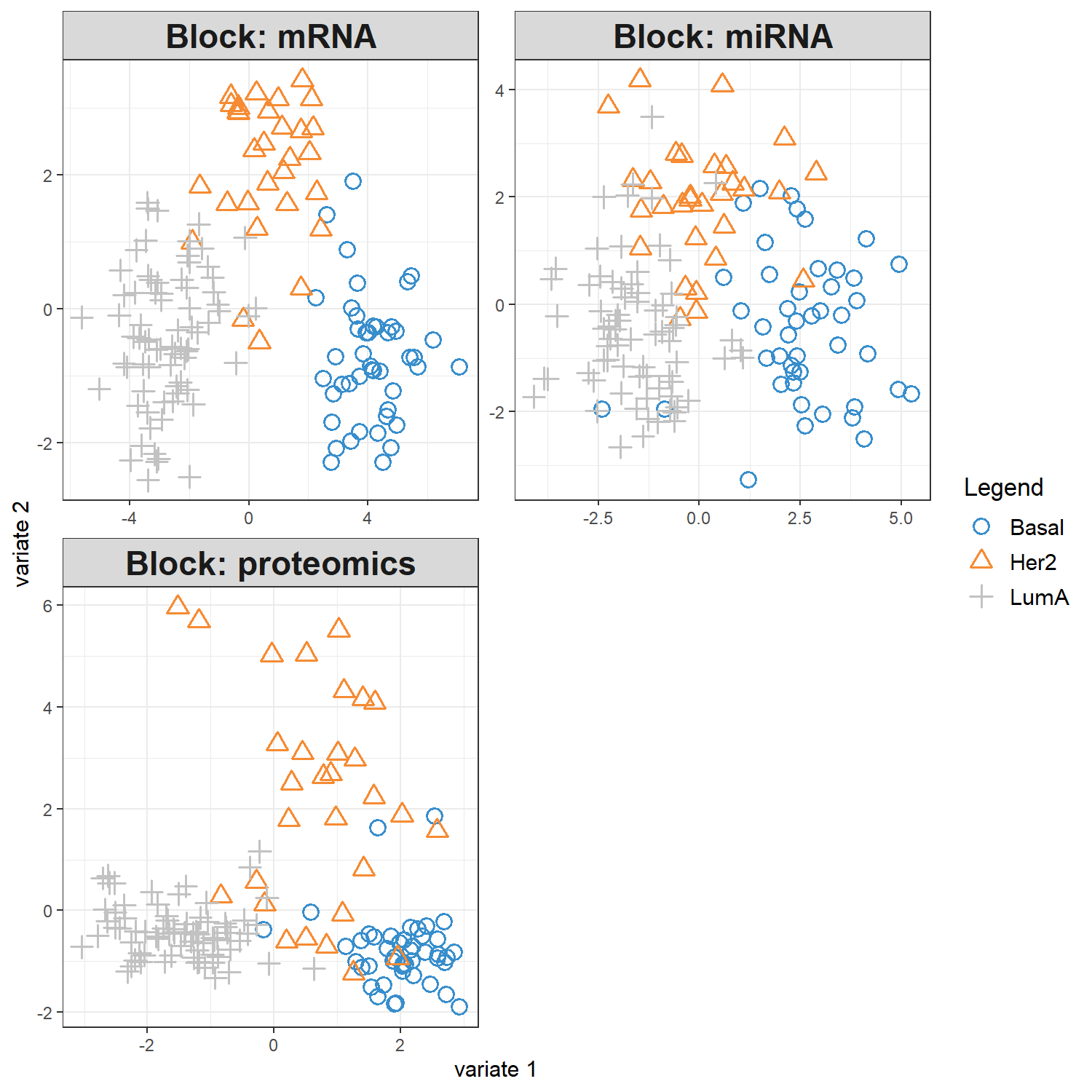
10.3 plotArrow
plotArrow() 顯示 同一樣本 在不同 omics 空間中的投影位置,箭頭越短表示不同 omics 對該樣本的表示越一致。
Important目的
評估不同 omics 資料區塊對同一樣本的表示是否一致,並檢視多體學整合的效果。
Tip如何解讀 plotArrow 圖
在 plotArrow() 圖中:
1️⃣ 每條箭頭代表一個樣本 (sample)
箭頭的起點與終點分別代表該樣本在不同 omics block 的 component 空間位置。
2️⃣ 箭頭長度 (arrow length)
箭頭長度反映不同 omics 對該樣本的表示差異:
箭頭較短
→ 不同 omics 對該樣本的表現模式一致
→ 表示多體學整合效果良好箭頭較長
→ 不同 omics 對該樣本的表示差異較大
→ 可能表示某些 omics block 捕捉到不同的訊號
3️⃣ 顏色代表樣本分類 (group / phenotype)
例如本範例中的 Basal、Her2、LumA。
若同一分類的樣本箭頭方向相似且長度較短,通常表示:
- 不同 omics 在該分類中捕捉到一致的 biological signal
- DIABLO 在多體學整合上具有良好的表現
因此,plotArrow() 主要用於 評估不同 omics 對樣本表現的整合一致性。
png(filename = "./images/arrow.png", width = 1500, height = 1500, res = 200)
plotArrow(sgccda.res, ind.names = FALSE, legend = TRUE, title = 'DIABLO')
dev.off()![]()
10.4 plotVar
plotVar() 可用於觀察不同 omics 特徵在 component 空間中的分布與關聯。
Important目的
探索不同 omics 特徵在 component 空間中的關係,並觀察不同體學之間是否存在相關的 biomarker。
Tip如何解讀 plotVar 圖
在 plotVar() 圖中:
1️⃣ 每個點代表一個特徵 (feature)
例如:
- mRNA:gene
- miRNA:microRNA
- proteomics:protein
這些特徵的位置由其在 component 空間中的 loading 決定。
2️⃣ 顏色代表不同 omics block
不同顏色用來區分不同資料區塊,例如:
- mRNA
- miRNA
- proteomics
3️⃣ 點之間的距離代表特徵之間的關聯
在 component 空間中:
- 距離越近 → 表示特徵具有較高相關性
- 距離較遠 → 表示特徵關聯較弱
若來自不同 omics 的特徵聚集在一起,通常表示這些分子可能參與相同的 biological process 或 regulatory network。
4️⃣ 方向代表與 component 的關聯
位於同一方向的特徵通常具有相似的變化模式,而方向相反的特徵則可能呈現負相關。
因此,plotVar() 主要用於 探索跨體學 biomarker 之間的潛在關聯與調控模式。
png(filename = "./images/var.png", width = 1500, height = 1500, res = 200)
plotVar(sgccda.res, var.names = FALSE, style = 'graphics', legend = TRUE,
pch = c(16, 17, 15), cex = c(2,2,2), col = c('darkorchid', 'brown1', 'lightgreen'))
dev.off()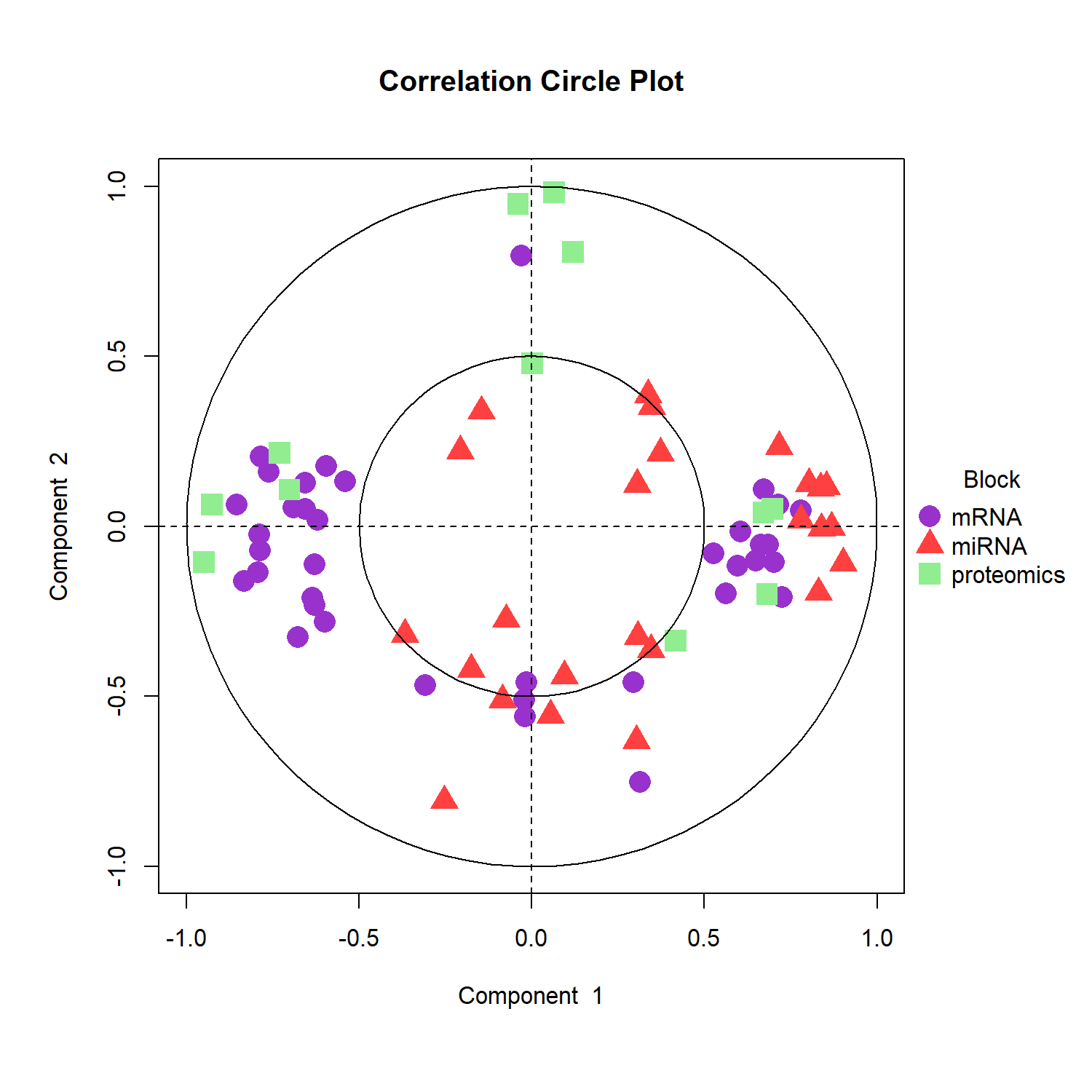
也可以只顯示特定 component 的變數分布,例如只觀察 component 1 的特徵關係。
png(filename = "./images/var-comp.png", width = 1500, height = 1500, res = 200)
plotVar(sgccda.res, var.names = FALSE, style = 'graphics', legend = TRUE,
pch = c(16, 17, 15), cex = c(2,2,2), col = c('darkorchid', 'brown1', 'lightgreen'),
comp.select = 1)
dev.off()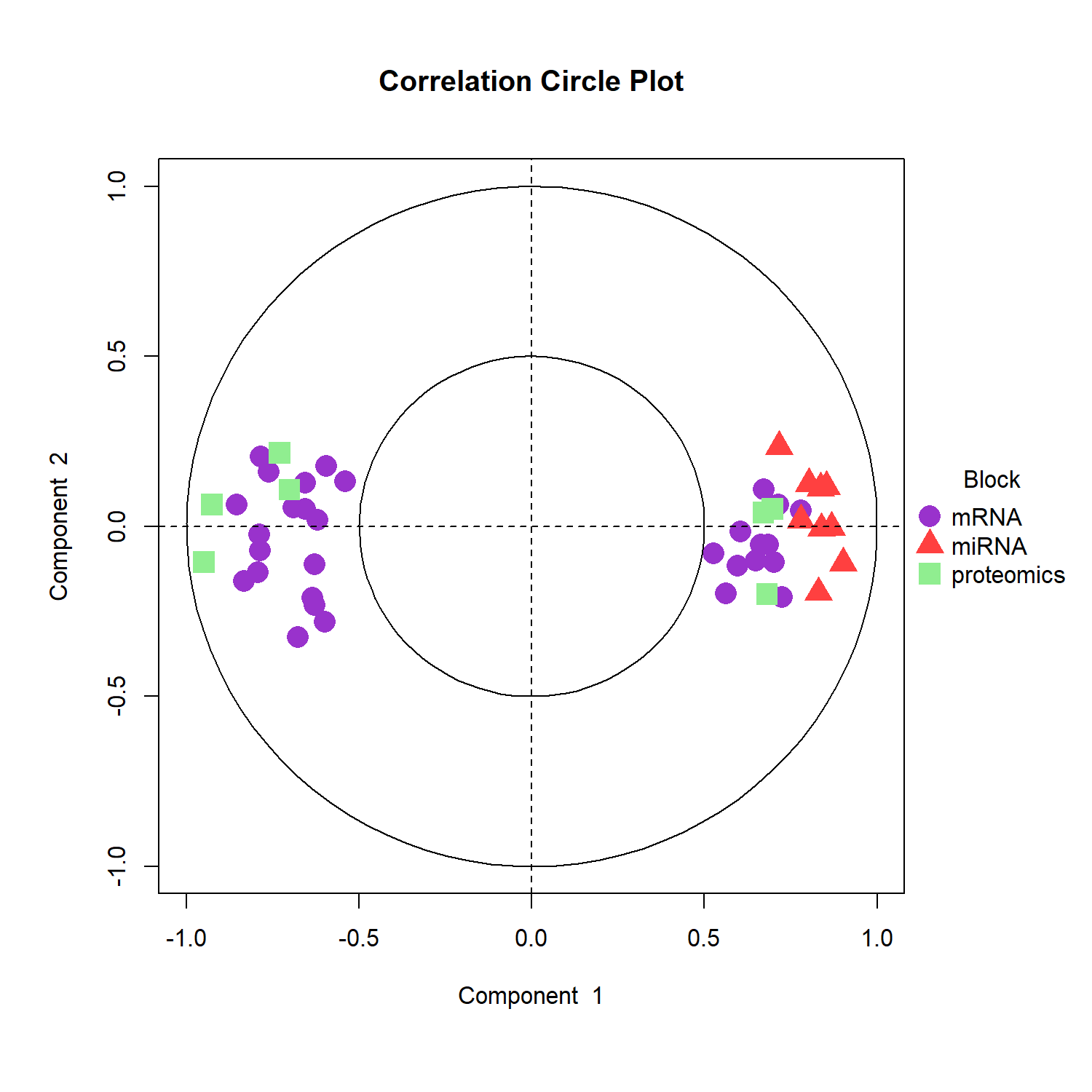
10.5 circosPlot
circosPlot() 以 circos diagram 的方式顯示 不同 omics 特徵之間的相關性,常用於探索跨體學的分子關聯。
Important目的
視覺化不同 omics block 之間所選出的 biomarker,並觀察跨體學特徵之間的相關性。
Tip如何解讀 circosPlot 圖
在 circosPlot() 圖中:
1️⃣ 圓形區塊 (blocks)
圓周上的區塊代表不同的 omics 資料,例如:
- mRNA
- miRNA
- proteomics
每個區塊中排列的是 DIABLO 模型所選出的特徵(biomarker)。
2️⃣ 圓弧線條 (links)
不同區塊之間的線條代表特徵之間的相關性:
- 紅色 / 深色線條 → 正相關
- 灰色 / 深色線條 → 負相關
只有相關係數高於 cutoff(本例為 0.7)的特徵關係才會顯示。
3️⃣ 跨體學關聯 (cross-omics links)
若 mRNA、miRNA 與 proteomics 之間存在大量連線,通常表示:
- 不同 omics 層級之間存在強烈的分子關聯
- 可能形成跨體學的 regulatory network
因此,circosPlot() 常用於探索 multi-omics biomarker network,並協助理解不同分子層級之間的潛在調控關係。
png(filename = "./images/circos.png", width = 1500, height = 1500, res = 200)
circosPlot(sgccda.res, cutoff = 0.7, line = TRUE,
color.blocks= c('darkorchid', 'brown1', 'lightgreen'),
color.cor = c("chocolate3","grey20"), size.labels = 1.5)
dev.off()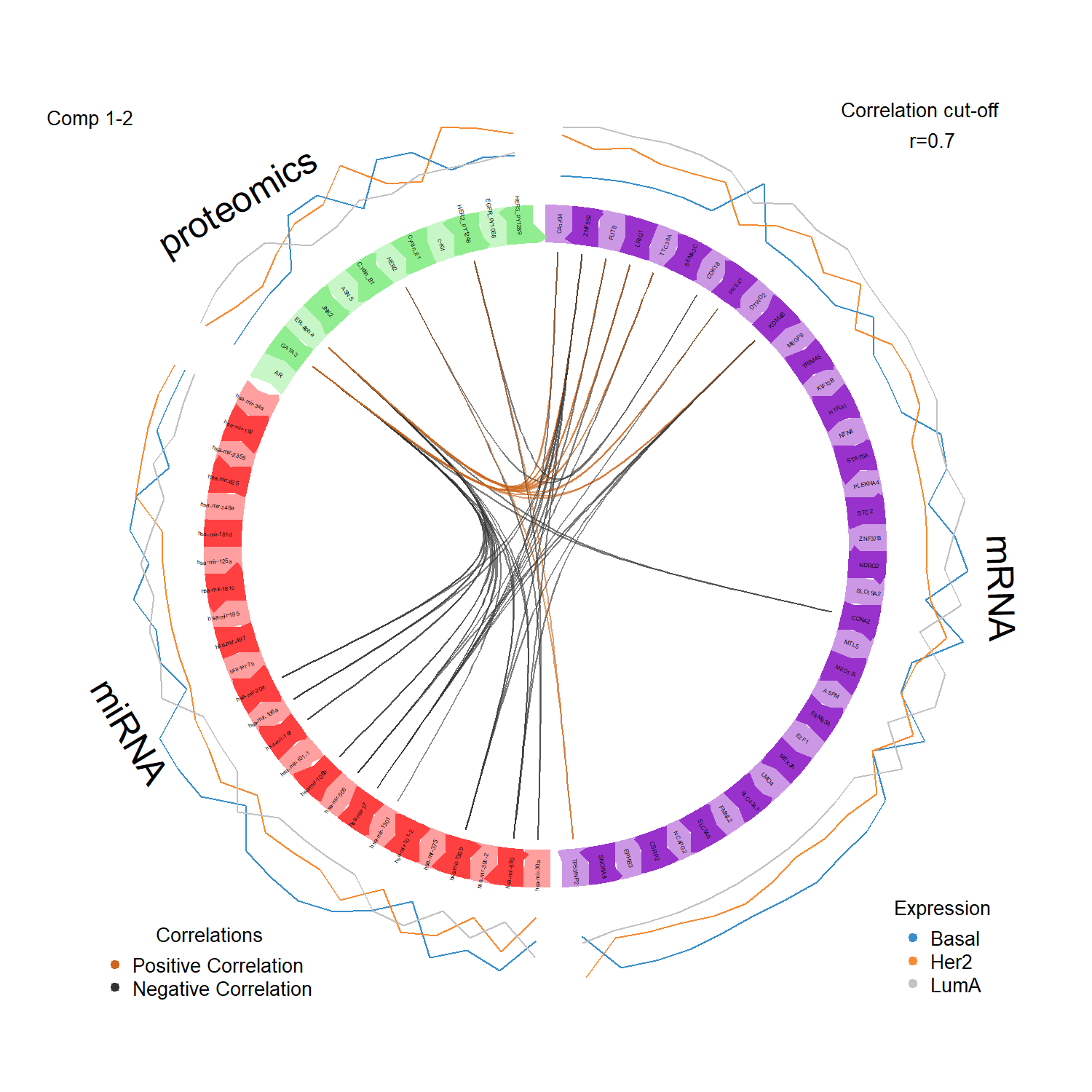
10.6 network
network() 則可建立特徵之間的網路結構,顯示不同 omics biomarker 之間的相關性。
Important目的
將 DIABLO 模型所選出的多體學特徵轉換為網路結構,以探索不同 omics biomarker 之間的潛在分子關聯。
Tip如何解讀 network 圖
在 network() 圖中:
1️⃣ 節點 (nodes)
每個節點代表一個被 DIABLO 選出的特徵,例如:
- gene(mRNA)
- miRNA
- protein
節點顏色代表不同的 omics block,例如:
- 紫色 → mRNA
- 棕色 → miRNA
- 綠色 → proteomics
2️⃣ 連線 (edges)
節點之間的連線代表特徵之間的相關性:
- 若兩個特徵的相關係數高於設定的
cutoff(本例為 0.4),則會建立連線。 - 連線越多,表示該特徵可能與更多分子具有相關關係。
3️⃣ 網路結構 (network structure)
在網路圖中:
高度連接的節點 (hub nodes)
→ 可能代表重要的 regulatory molecule跨體學連線 (cross-omics links)
→ 可能代表不同分子層級之間的調控關係
例如 miRNA–mRNA 或 gene–protein interaction。
因此,network() 可用於探索 multi-omics biomarker network,並協助發現潛在的關鍵調控分子。
png(filename = "./images/network.png", width = 1500, height = 1500, res = 200)
network(sgccda.res, blocks = c(1,2,3),
color.node = c('darkorchid', 'brown1', 'lightgreen'), cutoff = 0.4)
dev.off()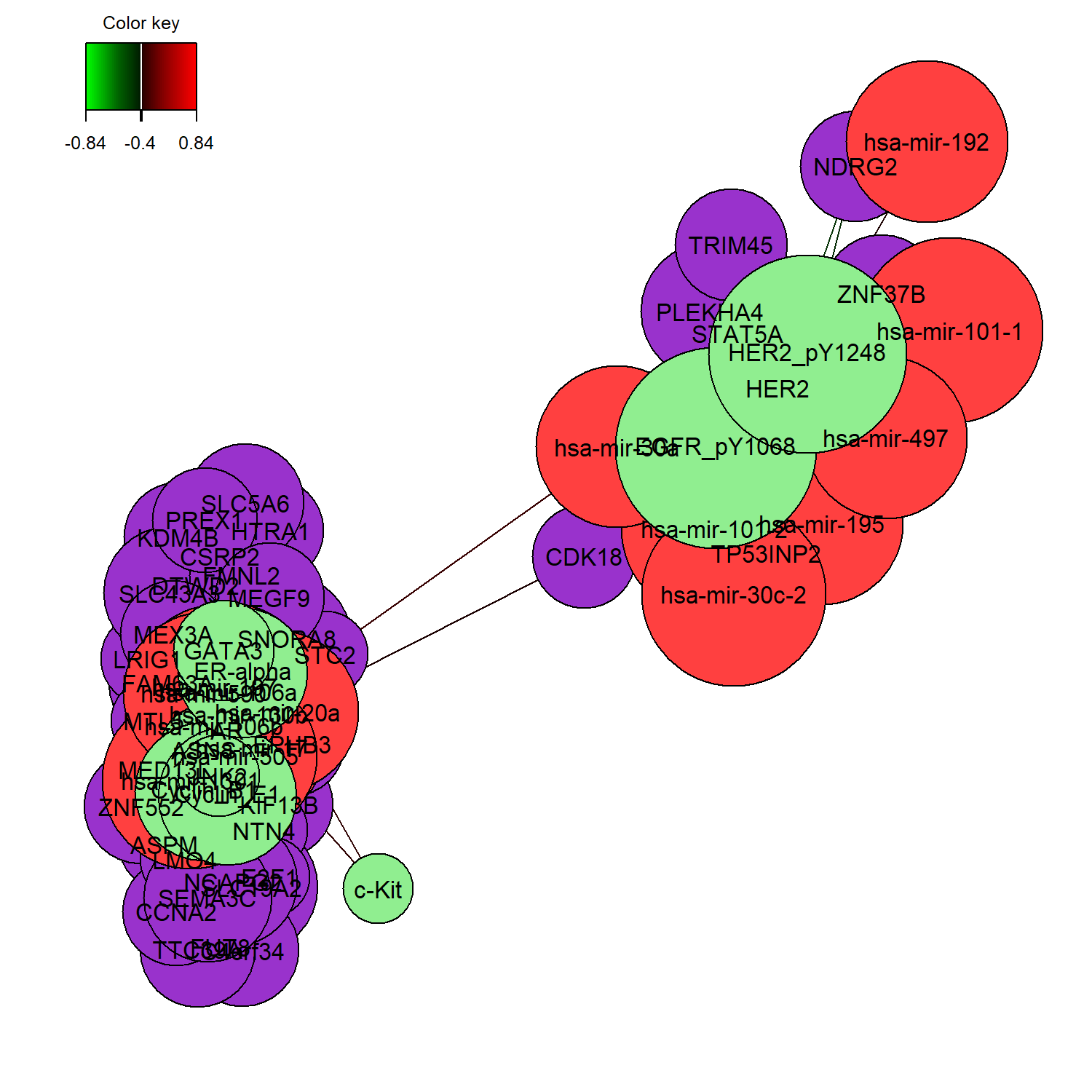
若需要進一步進行網路分析,可將 network 匯出為 GML 格式,並於 Cytoscape 或其他網路分析工具中進行視覺化。
library(igraph)
my.network <- network(sgccda.res, blocks = c(1,2,3),
color.node = c('darkorchid', 'brown1', 'lightgreen'), cutoff = 0.4)
write_graph(my.network$gR, file = "data/myNetwork.gml", format = "gml")10.7 plotLoadings
plotLoadings() 用於顯示 每個 component 中對模型貢獻最大的變數。
Important目的
識別在特定 component 中對模型貢獻最大的特徵,並找出可能的重要 biomarker。
Tip如何解讀 plotLoadings 圖
在 plotLoadings() 圖中:
1️⃣ 每個長條代表一個特徵 (feature)
這些特徵是 DIABLO 在指定 component 中選出的變數,例如:
- gene(mRNA)
- miRNA
- protein
不同 panel 代表不同的 omics block。
2️⃣ 長條長度代表 loading 大小
loading 表示該特徵在 latent component 中的權重:
- |loading| 越大 → 對 component 的貢獻越大
- |loading| 越小 → 對 component 的影響較小
因此 loading 較大的特徵通常是模型中較重要的 biomarker。
3️⃣ 正負 loading 代表特徵與 component 的方向
正 loading
→ 與 component 呈正相關負 loading
→ 與 component 呈負相關
這表示該特徵在不同樣本群體中可能呈現相反的表現模式。
4️⃣ 顏色代表主要貢獻的 phenotype
顏色表示該特徵主要與哪一個分類群體相關,例如:
- 藍色 → Basal
- 橘色 → Her2
- 灰色 → LumA
表示該特徵在該 subtype 中具有較強的 discriminative contribution。
因此,plotLoadings() 可用於識別 在特定 component 中最具分類能力的 multi-omics biomarker。
png(filename = "./images/loadings.png", width = 1500, height = 1500, res = 200)
plotLoadings(sgccda.res, comp = 2, contrib = 'max', method = 'median')
dev.off()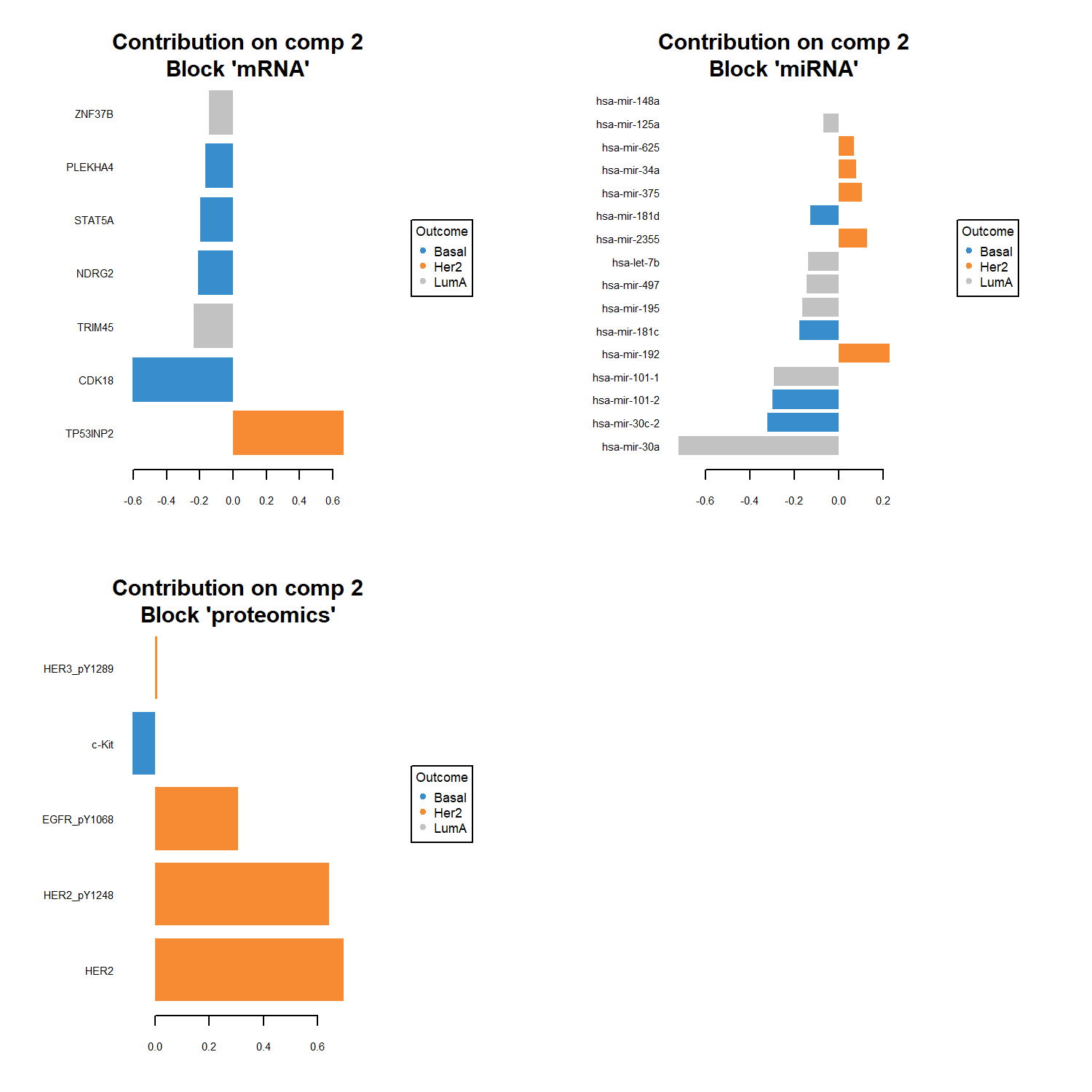
10.8 cimDiablo
最後,cimDiablo() 可以繪製 clustered image map (heatmap),顯示 不同 omics 特徵 在 樣本之間 的表現模式。
Important目的
整合不同 omics 所選出的特徵,並觀察這些 biomarker 在樣本之間的表現模式與群集關係。
Tip如何解讀 cimDiablo 圖
在 cimDiablo() 圖中:
1️⃣ 列 (rows) 代表樣本 (samples)
每一列代表一個樣本,左側的顏色條表示樣本的分類,例如:
- Basal
- Her2
- LumA
樣本通常會依據表現模式進行 hierarchical clustering,因此相似的樣本會被分在同一群。
2️⃣ 欄 (columns) 代表特徵 (features)
每一欄代表一個被 DIABLO 模型選出的特徵,例如:
- gene(mRNA)
- miRNA
- protein
上方的顏色條表示不同 omics block,例如:
- 紫色 → mRNA
- 紅色 → miRNA
- 綠色 → proteomics
3️⃣ 顏色代表表現量 (expression level)
熱圖中的顏色表示特徵在不同樣本中的相對表現:
- 紅色 / 暖色 → 較高表現
- 藍色 / 冷色 → 較低表現
通常資料會經過標準化(例如 z-score),因此顏色代表相對表現強度。
4️⃣ 樣本群集 (sample clustering)
若樣本依照 phenotype(例如癌症 subtype)形成明顯的群集,表示:
- 模型選出的特徵具有良好的分類能力
- 這些 multi-omics biomarker 能夠區分不同 biological states。
因此,cimDiablo() 常用於 整體檢視 multi-omics biomarker signature 在不同樣本之間的表現模式與群集關係。
png(filename = "./images/cimdiablo.png", width = 1500, height = 1500, res = 200)
cimDiablo(sgccda.res, color.blocks = c('darkorchid', 'brown1', 'lightgreen'),
comp = 1, margin=c(8,20), legend.position = "right")
dev.off()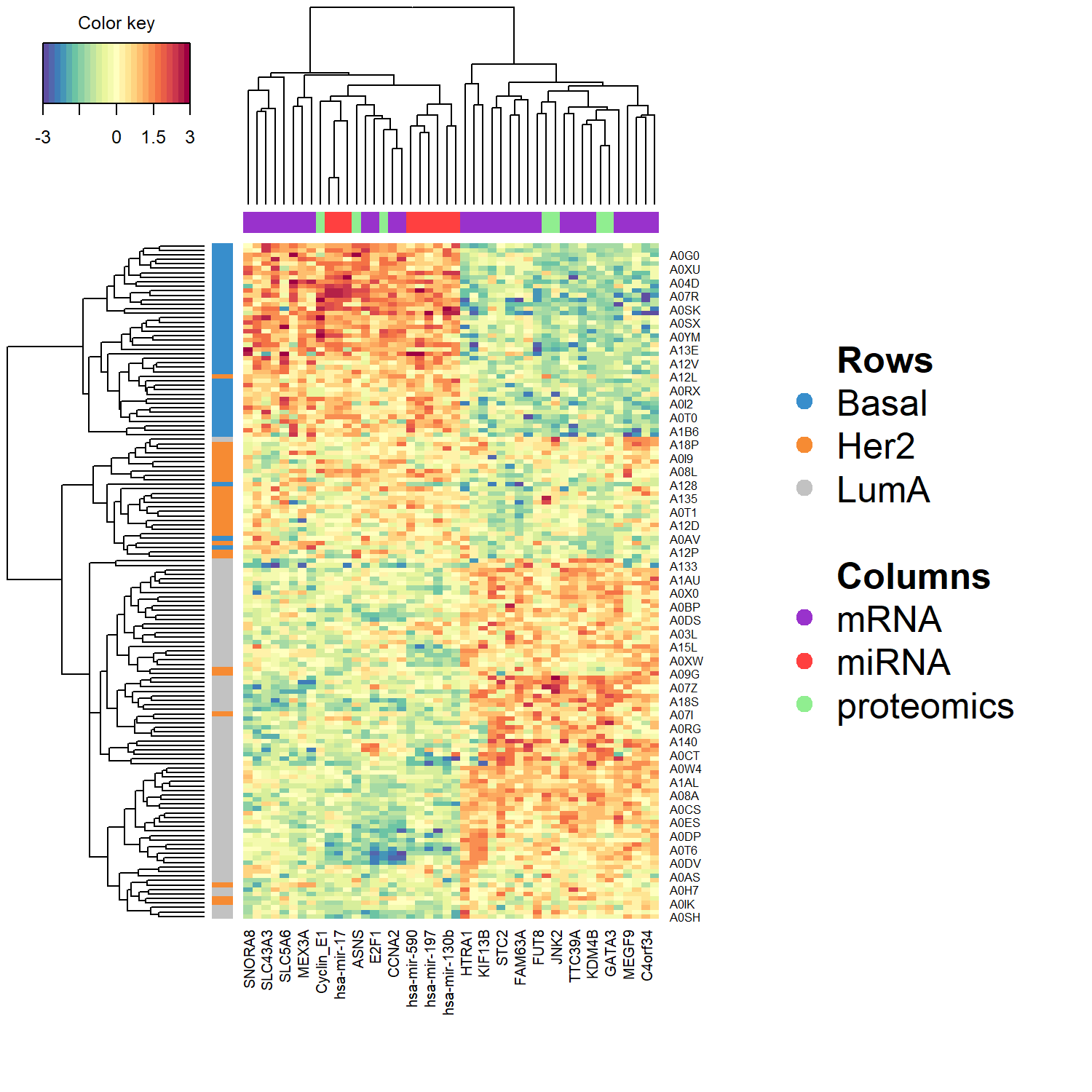
11 模型評估
在建立最終 DIABLO 模型後,可以透過 perf() 進行模型效能評估。本範例採用 10-fold cross-validation 並重複 10 次,以評估模型在不同資料分割下的分類表現。dist = “centroids.dist” 表示使用 centroid distance 作為分類判定方式。此外,利用 proc.time() 記錄模型評估所需的運算時間。
t1 <- proc.time()
perf.diablo <- perf(
sgccda.res,
validation = "Mfold",
folds = 10,
nrepeat = 10,
dist = "centroids.dist"
)
running_time <- proc.time() - t1
running_time user system elapsed
7.47 0.22 7.69 perf() 的結果可提供不同投票策略下的分類錯誤率,包括 Majority vote 與 Weighted vote。Majority vote 以多數決方式整合不同 omics block 的預測結果,而 Weighted vote 則會根據各 block 的預測表現進行加權。
perf.diablo$MajorityVote.error.rate
perf.diablo$WeightedVote.error.rate$centroids.dist
comp1 comp2
Basal 0.03333333 0.03333333
Her2 0.17333333 0.14000000
LumA 0.06533333 0.01600000
Overall.ER 0.07733333 0.04600000
Overall.BER 0.09066667 0.06311111
$centroids.dist
comp1 comp2
Basal 0.01555556 0.03333333
Her2 0.11666667 0.10666667
LumA 0.06533333 0.01466667
Overall.ER 0.06066667 0.03866667
Overall.BER 0.06585185 0.05155556此外,也可以使用 auroc() 計算 ROC curve 與 AUC (Area Under the Curve),用來評估模型的分類能力。此範例以 miRNA block 的第二個 component 為例計算 AUC。
png(filename = "./images/auroc.png", width = 1500, height = 1500, res = 200)
auc.diablo <- auroc(sgccda.res, roc.block = "miRNA", roc.comp = 2)
dev.off()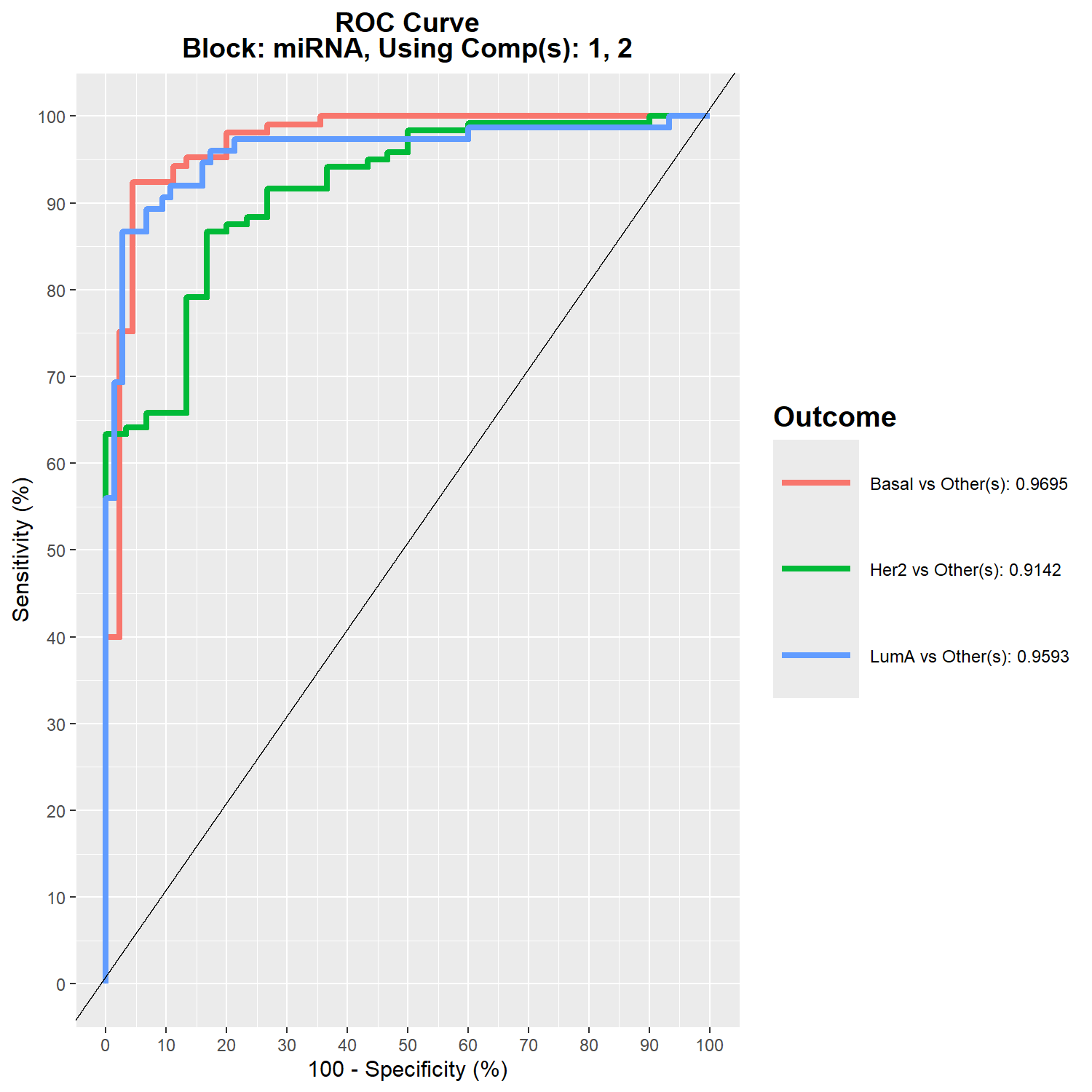
12 外部驗證
為了評估模型在新資料上的預測能力,可以使用 獨立的測試資料(test set) 進行外部驗證。本範例使用 breast.TCGA 資料集中提供的 data.test 作為測試資料。此測試資料僅包含 mRNA 與 miRNA 兩個 omics block,而缺少 proteomics block,DIABLO 模型仍可利用現有的資料區塊進行預測。
data.test.TCGA <- list(
mRNA = breast.TCGA$data.test$mrna,
miRNA = breast.TCGA$data.test$mirna
)
predict.diablo <- predict(
sgccda.res,
newdata = data.test.TCGA
)接著將模型預測結果與真實分類進行比較,建立 confusion matrix,並計算 Balanced Error Rate (BER) 來評估模型的分類表現。
confusion.mat <- get.confusion_matrix(
truth = breast.TCGA$data.test$subtype,
predicted = predict.diablo$WeightedVote$centroids.dist[, 2]
)
confusion.mat
get.BER(confusion.mat) predicted.as.Basal predicted.as.Her2 predicted.as.LumA
Basal 20 1 0
Her2 0 14 0
LumA 0 1 34
[1] 0.02539683Confusion matrix 可顯示不同分類之間的預測結果,而 BER 則可在類別數量不平衡的情況下提供較公平的分類評估指標。